pipelines-docs
Phasing Workflow
The workflow and documentation were developed and executed by David Tang (Po-Ru Loh group) and collaborators.
Overview
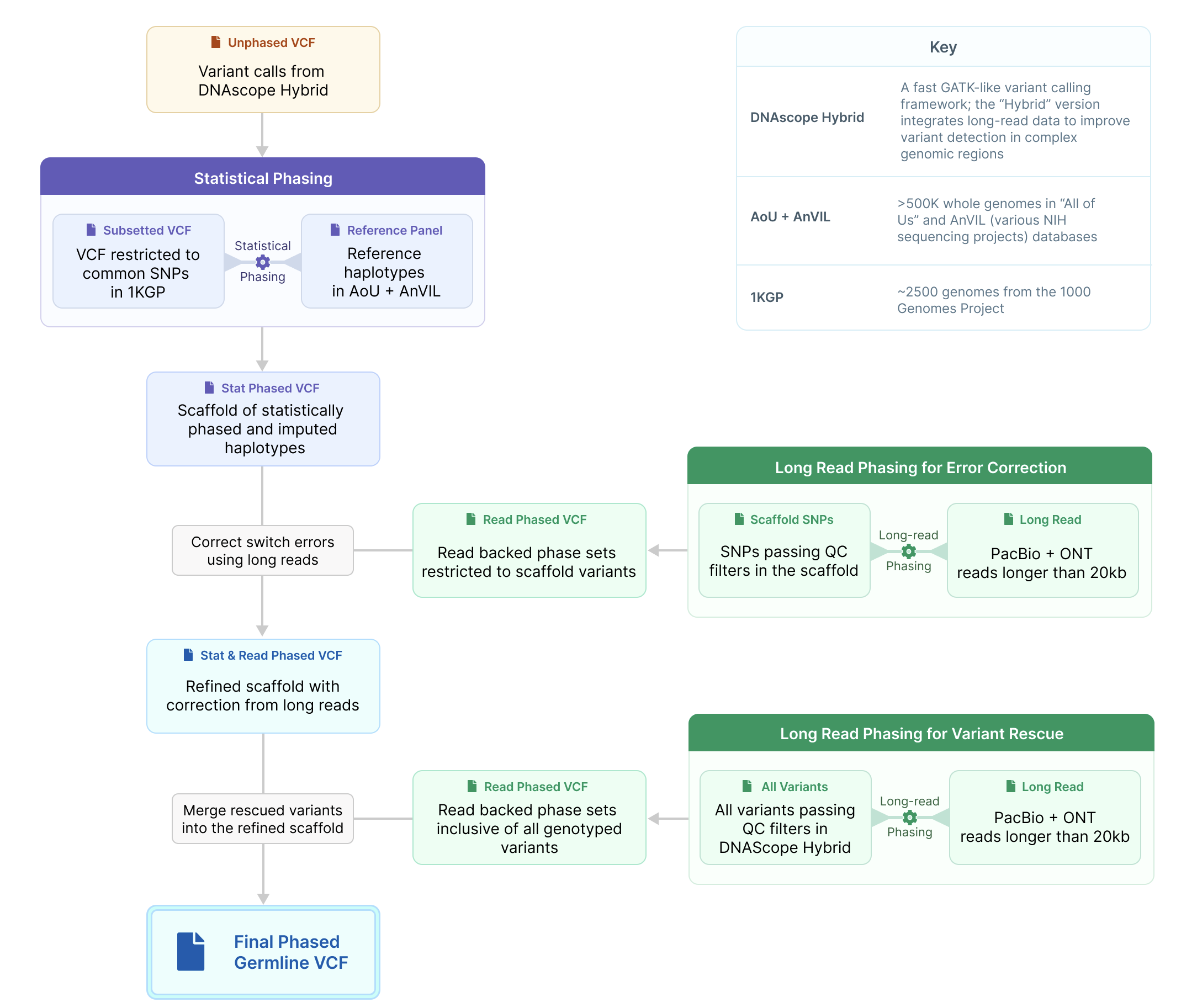
This workflow describes a strategy for generating high-quality phased VCFs by integrating statistical phasing (All of Us + AnVIL imputation server) with long-read phasing (PacBio + ONT phased using WhatsHap).
Strategy for combining long-read phasing with statistical phasing
1. Generate scaffold using statistical phasing
Statistical phasing leverages haplotype sharing in large reference panels to accurately infer long-range phase. We generated a statistically phased scaffold by passing common SNPs in the 1000 Genomes Project – genotyped in SMaHT – to the AoU+AnVIL Imputation Server, which imputes 989,868,760 variants genotyped in 515,579 ancestrally diverse genotypes across the All-of-Us and AnVIL datasets. We restricted the phased scaffold to correctly imputed sites, including both SNPs and indels.
2. Correct switch errors in the phased scaffold using long reads
Long reads offer an orthogonal approach to phasing by using physical linkage of alleles on the same read to phase nearby heterozygous sites. Unlike statistical phasing, which can produce long haplotypes accurate to 10Mb or more, read-backed phasing produces shorter phase sets (groups of variants that can be phased together) that get broken by runs of homozygosity which lack phaseable heterozygous sites. Phase sets are typically 1Mb or shorter, but can be highly accurate if restricted to high quality variant calls. Thus, phase sets provide an opportunity to correct errors in the scaffold.
The following outlines our approach to refine the phased scaffold using long reads.
- Phase only SNPs on the scaffold passing QC filters using WhatsHap (v2.8), with the
--distrust-genotypesflag enabled, on all reads (both PacBio and ONT from any tissue) longer than 20kb, with a mapping quality greater than 40, and SAM flags 3844 unset. We subsetted to only reads longer than 20kb for computational efficiency as the combined coverage across tissues afforded high coverage even when restricted to the longest, most phase informative reads (over ~100x coverage for most donors) - For each pair of consecutive variants in the same phase set, set the relative phase of the variants in the phase scaffold to the match the relative phase of variants in the read based phasing
- Leave the relative phase of other variants – those not contained in a phase set or those spanning the boundaries of phase sets – unchanged
This strategy can also be thought of as using the statistically phased scaffold to bridge between read-backed phase sets with the rationale that read-backed phasing gives highly accurate (but fragmented) phase sets.
3. Phase non-scaffold variants onto the refined scaffold
The phased scaffold is limited to variants in the AoU+AnVIL reference panel, which only consists of variants in regions mappable by short reads and typically represent ~2.4 million out of the ~3 million total heterozygous sites per genome. We phased an additional ~300,000 variants per genome onto the scaffolds as follows:
- Phase all SNPs and indels passing QC filters using WhatsHap (v2.8), with the
--distrust-genotypesflag enabled, on all reads (both PacBio and ONT from any tissue) longer than 20kb, with a mapping quality greater than 40, and SAM flags 3844 unset - Correct errors in the read-backed phasing using the refined scaffold (trusting the scaffold phase anytime the relative phase of two variants disagree)
- Remove phase from variants located between sites in the scaffold identified to harbour a switch error
Running read based phasing two separate times is necessary as incorrect genotype calls (or calls in difficult genomic regions supported by fewer reads) can increase the frequency of switch errors within phase sets. Using a high quality scaffold alleviates issues introduced by lower confidence variants by anchoring the phase against high confidence variants.
Benchmarking phase accuracy
High cell fraction mosaic chromosomal alterations provide an opportunity to assess the number of switch errors since they leave signatures of consistent allelic imbalance favoring the same haplotype (see Loh et al. 2018 Nature). We evaluated the number of switch errors using ~40 chromosomes with large mosaic chromosomal alterations of a moderate cell fraction (length > 50Mb and BAF deviation > 0.01). Using this set of mosaic chromosomal alterations, we verified that the statistical phasing alone produced haplotypes with a median of 1 switch error per ~10Mb, while the long read refinement reduced the median number of switch errors to 1 switch per ~30Mb.
Output files
The final phased VCFs are annotated with a PS FORMAT tag (populated by WhatsHap) to indicate sets of variants phased together using the long-reads. The VCFs also include a PHASE_SCAFFOLD INFO tag to indicate variants on the scaffold generated with statistical phasing.