pipelines-docs
Short-Read RNA-seq, Paired-End
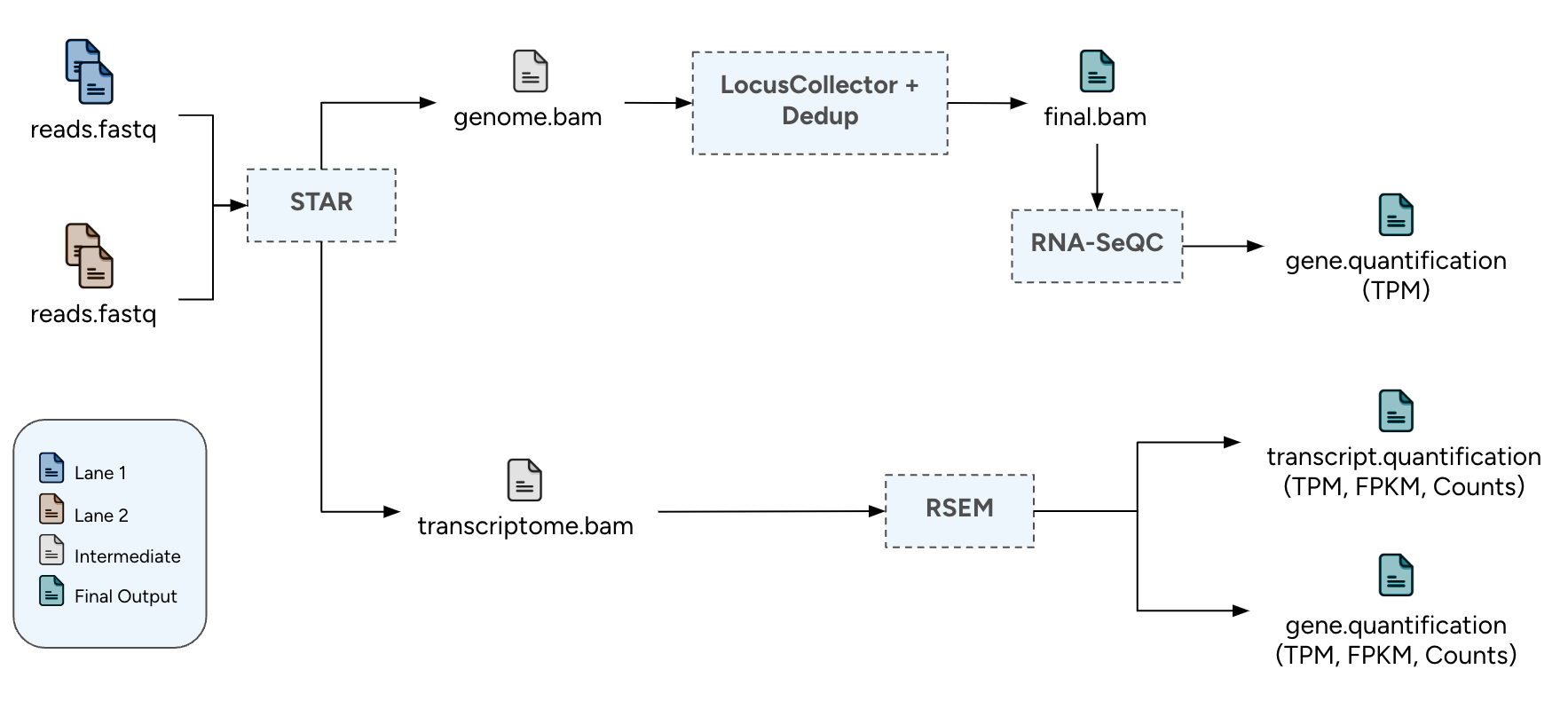
The paired-end short-read analysis pipeline for RNA-seq data is based on GTEx1 and TOPMed2 analysis pipelines. It is designed for per-sample execution, handling one or multiple sets of paired FASTQ files.
Key Pipeline Steps
- Alignment with STAR: Initial alignment of the raw reads to the reference genome and the transcriptome using STAR.
- Duplicate Reads Marking: Identification and labeling of duplicate reads, that originated during library preparation or as sequencing artifacts.
- Transcript Quantification: Transcript-level quantification using RSEM.
- Gene Quantification: Gene-level quantification using RNA-SeQC.
Sentieon Software
To meet the scalability demands, the pipeline utilizes a more efficient software implementation from Sentieon. Sentieon offers a comprehensive toolkit that replicates the original STAR and GATK algorithms, while enhancing computational efficiency.
Pipeline Chart
1: Lonsdale, J., Thomas, J., Salvatore, M. et al. The Genotype-Tissue Expression (GTEx) project. Nat Genet 45, 580–585 (2013). doi: 10.1038/ng.2653; 2: Taliun, D., Harris, D.N., Kessler, M.D. et al. Sequencing of 53,831 diverse genomes from the NHLBI TOPMed Program. Nature 590, 290–299 (2021). doi: 10.1038/s41586-021-03205-y